nadia-process
Overview
Perform data processing for single-cell/single-nucleus RNA-seq analysis.
nadia-process takes a matrix from nadia-quant and perform data
processing steps. We use Scanpy
package for most steps. They includes:
Filter cells by QC metrics
Filter genes
Doublet detection
Normalization
Feature selection (detect highly variable genes)
Dimentionality reduction (PCA)
Visualization (UMAP, T-SNE)
Users could re-run nadia-process several times to choose approriate parameters.
See: Single cell RNA-seq analysis workshop for more details.
How it works
1. Filter cells by QC metrics
There are some QC plots in the output of nadia-quant, which we can use to
choose threshold to filter cells.
We should remove cells with: * high proportion of mitochondrial count * low proportion of ribosomal count * too high or too low gene count * too high or too low UMI count
See Cell filter options for more details.
2. Filter genes
There are some options to filter genes from the matrix.
We can remove genes which do not meet the minimum number of cells expressed. See –min-cell option.
We can remove mitochondrial and ribosomal genes, as their expression is mainly technical problem.
Users can provides a list of gene symbols to remove by –remove-genes option.
3. Doublet detection
Doublets detection is performed by scrublet package. It is done after filter cells and genes. Most doublet detectors simulates doublets by merging cell counts and predicts doublets as cells that have similar embeddings as the simulated doublets. It requires users to input the expected doublet rate (–expected-rate option, default 0.06).
For each barcode, scrublet will produce 2 values:
Doublet score (doublet_scores column): higher value is more likely a barcode is doublet.
Prediction (predicted_doublets column): after calculating doublet score, scrublet automatically choose a threshold to classify barcode into singlet and doublet.
If automatic threshold detection doesn’t work well, you can adjust the threshold with –min-doublet-score option.
We also create some Doublet detection plots
Finally, if –filter-doublet flag is used, then predicted doublets will be removed.
4. Dimentionality reduction
Before variable gene selection we need to normalize and logaritmize the data.
# normalize to depth 10 000
sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
# logaritmize
sc.pp.log1p(adata)
Next, we need to define which features/genes are important in our dataset to distinguish cell types. For this purpose, we need to find genes that are highly variable across cells, which in turn will also provide a good separation of the cell clusters.
# compute variable genes
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# subset for variable genes in the dataset
adata = adata[:, adata.var['highly_variable']]
Since each gene has a different expression level, it means that genes with higher expression values will naturally have higher variation that will be captured by PCA. This means that we need to somehow give each gene a similar weight when performing PCA (see below). The common practice is to center and scale each gene before performing PCA.
Additionally, we can use regression to remove any unwanted sources of variation from the dataset, such as n_counts’, n_genes, ‘Mito_percent’, ‘Ribo_percent’,… Users can specify the list of variables to regress by –regress option.
# regress out unwanted variables
sc.pp.regress_out(adata, regress_variables)
# scale data, clip values exceeding standard deviation 10.
sc.pp.scale(adata, max_value=10)
Finally, we perform PCA and create Dimentionality reduction plots.
# PCA
sc.tl.pca(adata, svd_solver='arpack')
5. Visualization
The UMAP implementation in SCANPY uses a neighborhood graph as the distance
matrix, so we need to first calculate the graph. There are two available
parameters users could specify: number of principal components to use (–n-pcs),
and number of neighbors (–n-neighbors). We should choose the PCs contributing
most of variance by seeing PCA variance ratio plot. To learn about --n-neighbors,
click here.
sc.pp.neighbors(adata, n_pcs = n_pcs, n_neighbors = n_neighbors)
sc.tl.umap(adata)
We also run BH-tSNE.
sc.tl.tsne(adata, n_pcs = n_pcs)
Some metadata of cells are visualized in UMAP plot, TSNE plot and Violin plot (Metadata plots). If you want to see the expression level of genes of interest in UMAP plot, you can provide the list of gene symbol in –plot-genes argument (Gene of interest plot).
Input
Gene expression matrix in either mtx format (–mtx) or h5ad format (–h5ad).
Output
Processed AnnData object
Processed AnnData object is output in h5ad format. This file could be used for
further downstream analyses, such as sample integration by nadia-combine,
visualization by cellxgene,…
Report
nadia-process produces a multiqc report in html format. You can download an
example report
Plots
Doublet detection plots
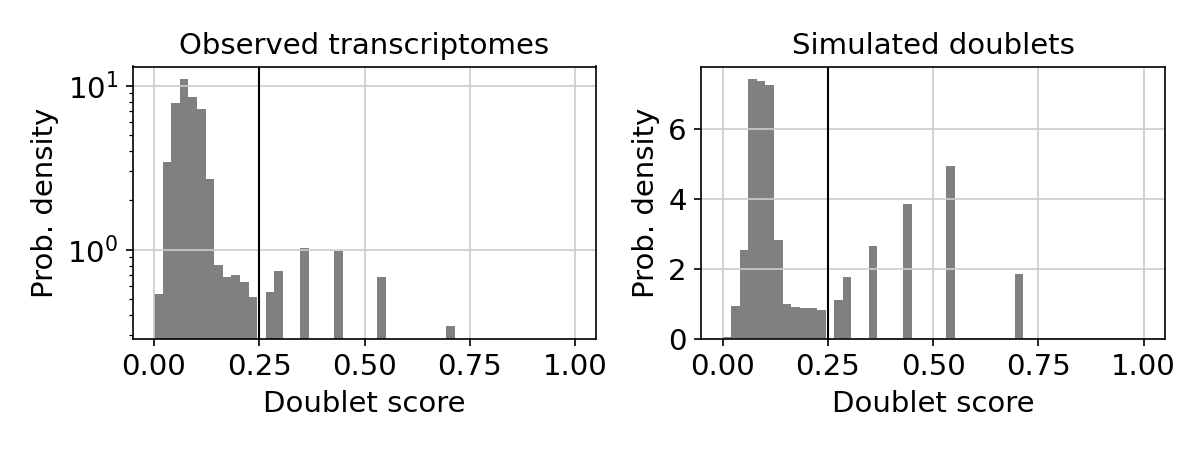
Doublet score histogram
This plot shows the doublet score histograms of observed transcriptomes and simulated doublets. The vertical line is the doublet score threshold.
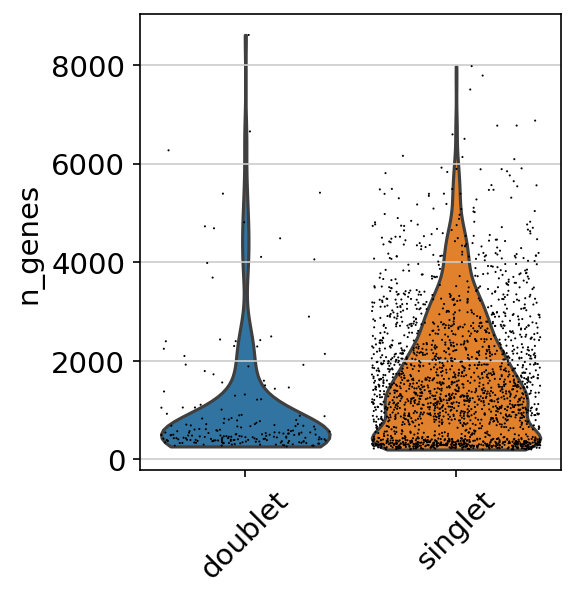
Doublet violin plot
This violin plot shows the difference in the number of genes between singlet and doublet. We can expect that doublets have more genes than singlets.
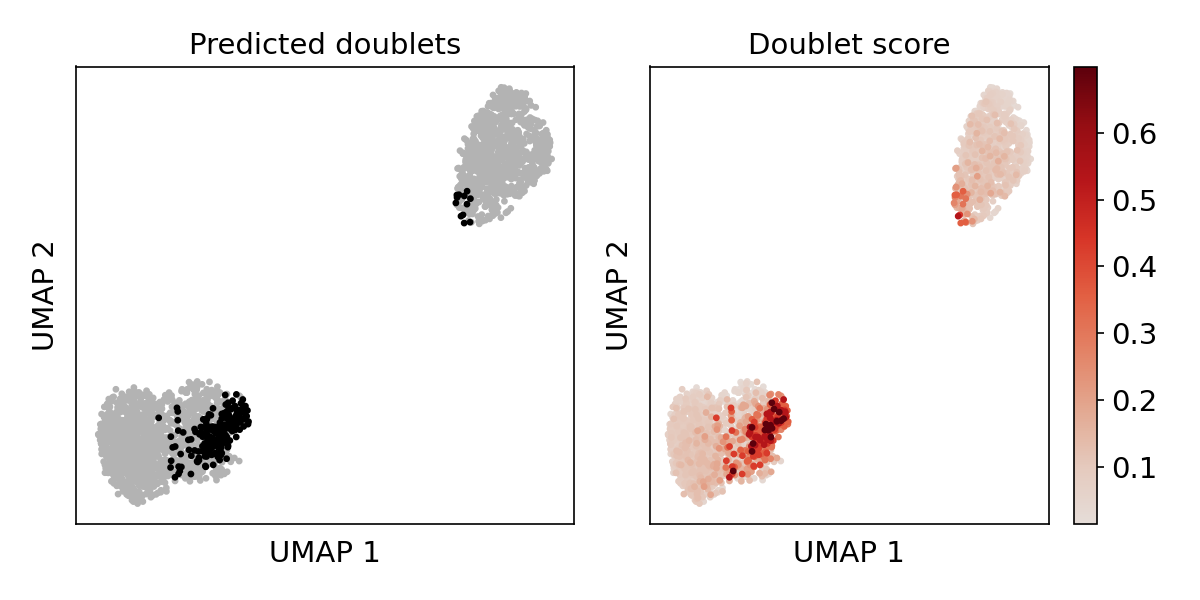
Doublet score UMAP plot
This plot shows the doublet score and the location of predicted doublet in UMAP embedding. Predicted doublets should co-localize in distinct states.
Dimentionality reduction plots
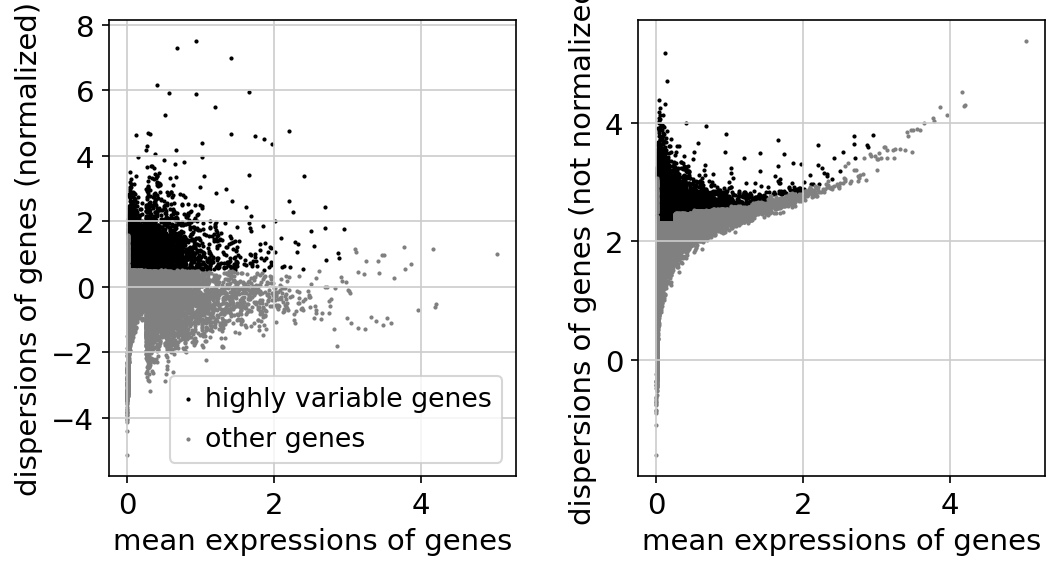
Gene dispersion plot
This plot shows the dispersion of genes by mean expression and shows genes that are filtered out.
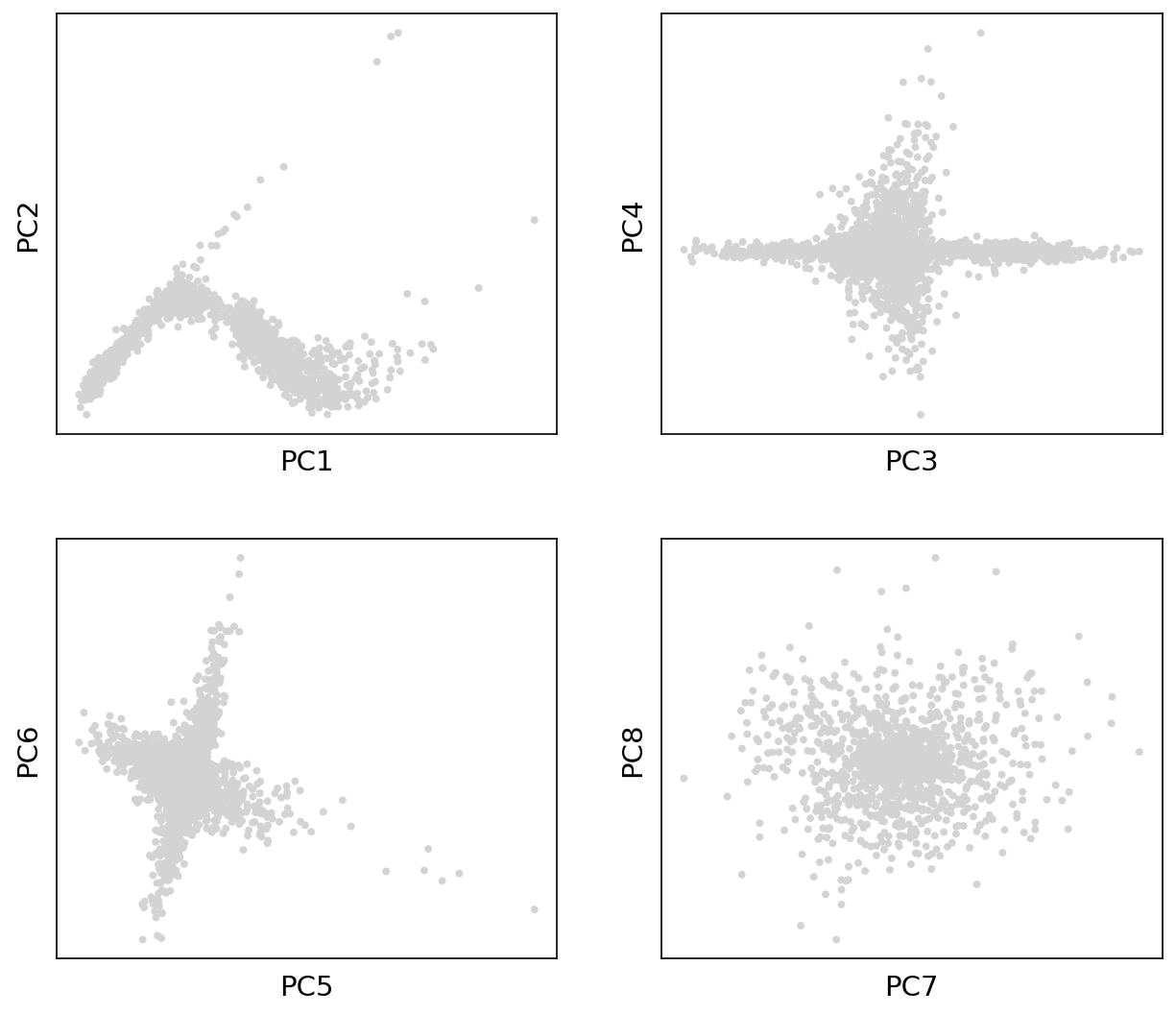
PCA plot
This plot shows the first principal components.
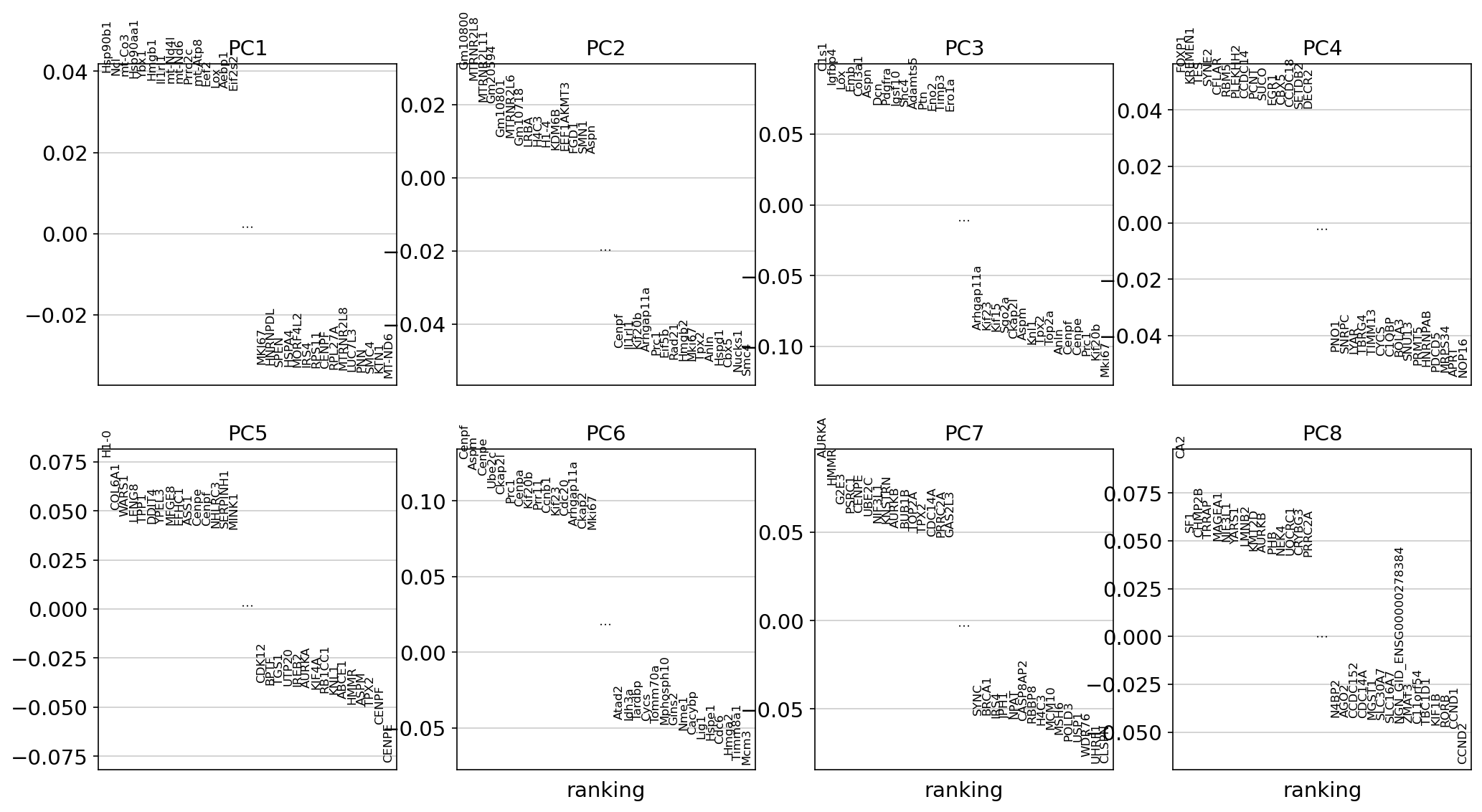
PCA loading plot
This plot identifies genes that contribute most to each PC, one can retrieve the loading matrix information.
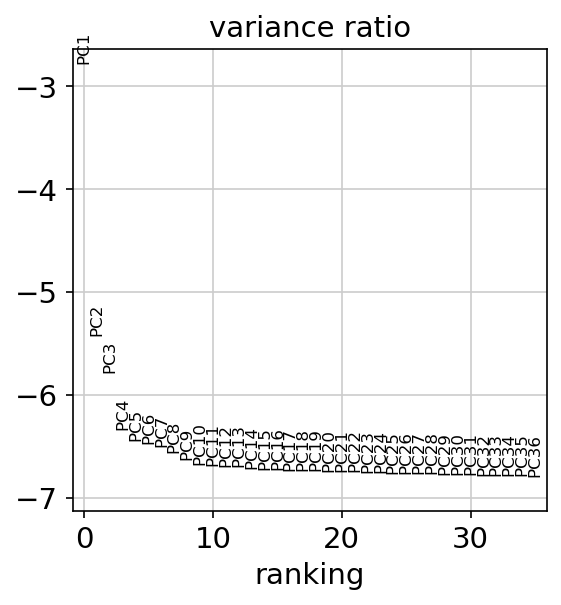
PCA variance ratio plot
This plot shows the amount of variance explained by each PC. This could be use to choose the number of PCs to use in the next steps.
Metadata plots
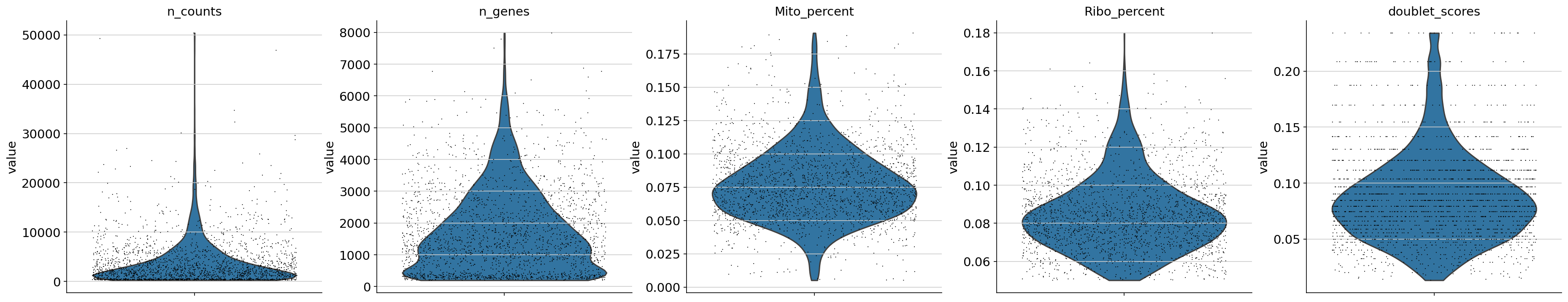
Violin metadata plot
This plot shows metadata after applying all filtering.
UMAP metadata plot
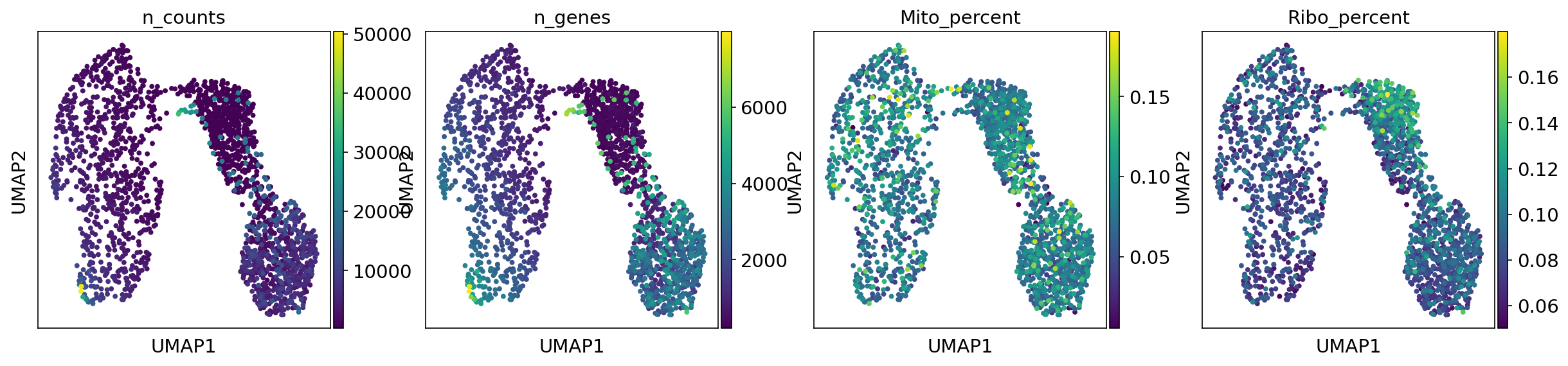
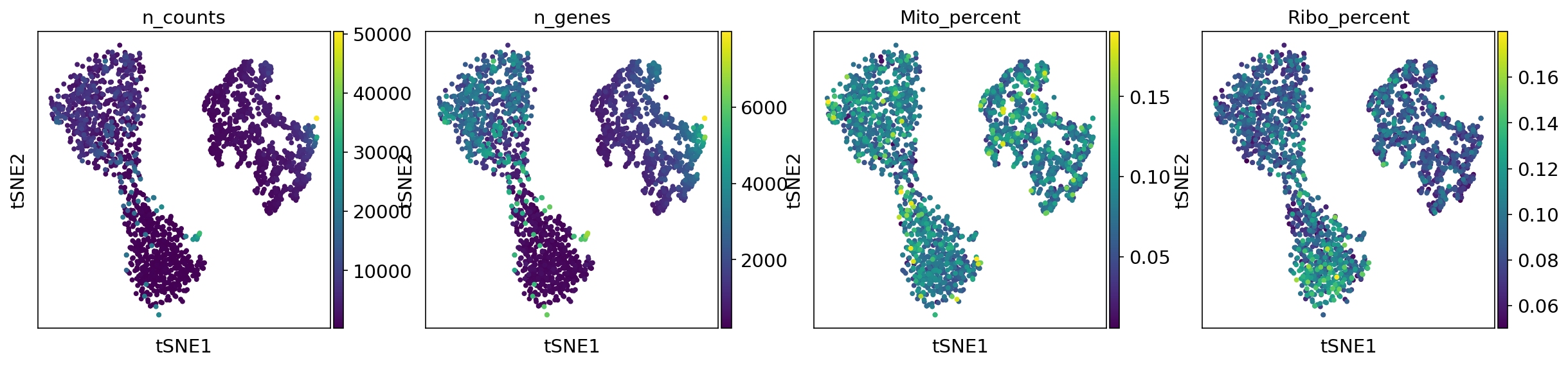
T-SNE metadata plot
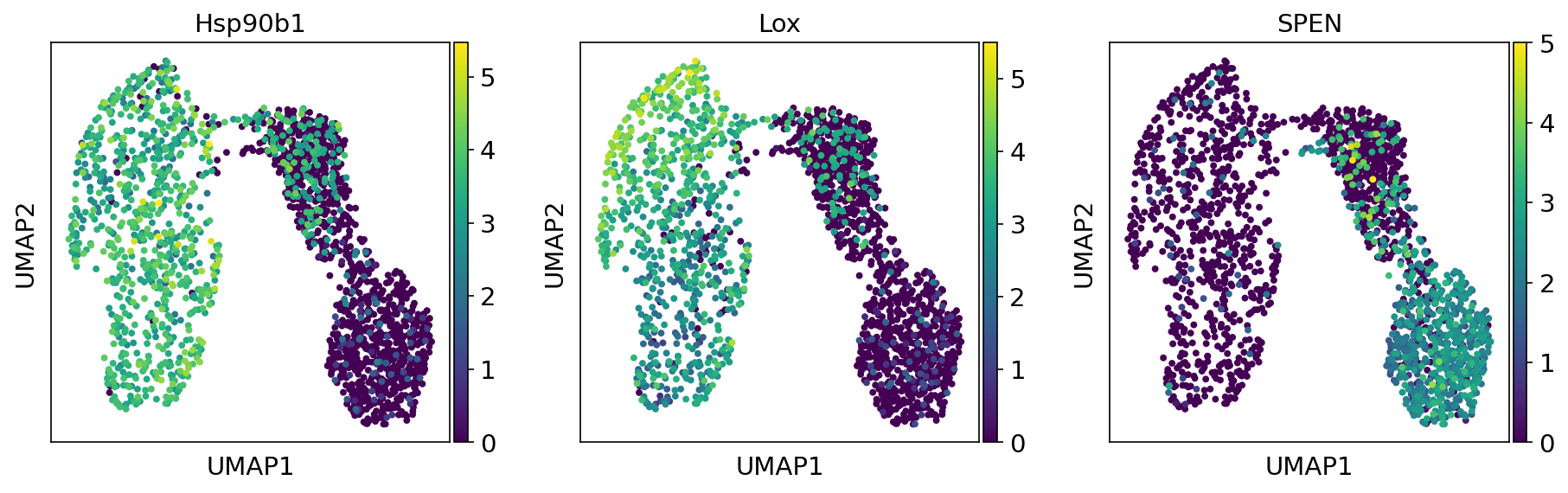
Gene of interest plot
Usage examples
Use h5ad file as input.
nadia-process \
--h5ad anndata/140922_SC_4_filter.h5ad \
-o nadiaprocess \
--filter-doublet \
--min-doublet-score 0.25 \
--n-pcs 10 \
--plot-genes Hsp90b1 Lox LOLC1 SPEN
Use mtx file as input.
nadia-process \
--mtx MTX/140922_SC_4/filter \
-o nadiaprocess \
--filter-doublet \
--n-pcs 10
Argument details
Input/Output options
--h5ad
Required
Path to h5ad file (AnnData object)
--mtx
Required
Path to matrix folder, which contains matrix.mtx, features.tsv, barcodes.tsv (gz support)
-o, --outdir
Required
Output directory
-n, --name
Sample name. It will be used for naming output files.
Required if input matrix is in mtx format. If not specified, then filename of h5ad file will be used for sample name.
Cell filter options
--mito
Default: “+MT-”
Regular Expression string of mitochondrial genes
--max-mito
Default: 0.2
Maximum percentage of mitochondrial genes.
--ribo
Default: “+RP[SL]”
Regular Expression string of ribosomal genes
--min-ribo
Default: 0.05
Minimum percentage of ribosomal genes
--min-gene
Default: 200
Minimum number of gene count
--max-gene
Maximum number of gene count
--min-umi
Minimum number of UMI count
--max-gene
Maximum number of UMI count
Doublet filter options
--filter-doublet
If this flag is used, filter doublets
--expected-rate
Default: 0.06
Expected doublet rate
--min-doublet-score
Default: None
Minimun doublet score. If None, select threshold automatically.
Gene filter options
--min-cell
Default: 3
Filter genes by number of cells.
--remove-mito
If this flag is used, remove mitochondrial genes
--remove-ribo
If this flag is used, remove ribosomal genes
--remove-genes
List of genes to remove (gene symbols)
Visualization options
--regress
Default: [‘n_counts’, ‘Mito_percent’]
List of variable to regress. Options: n_counts, n_genes, Mito_percent,…
--n-pcs
Default: 30
Number of Principle Components to compute UMAP and tSNE.
--n-neighbors
Default: 20
Number of neighbors to compute UMAP.
--plot-genes
List of genes of interest to plot.